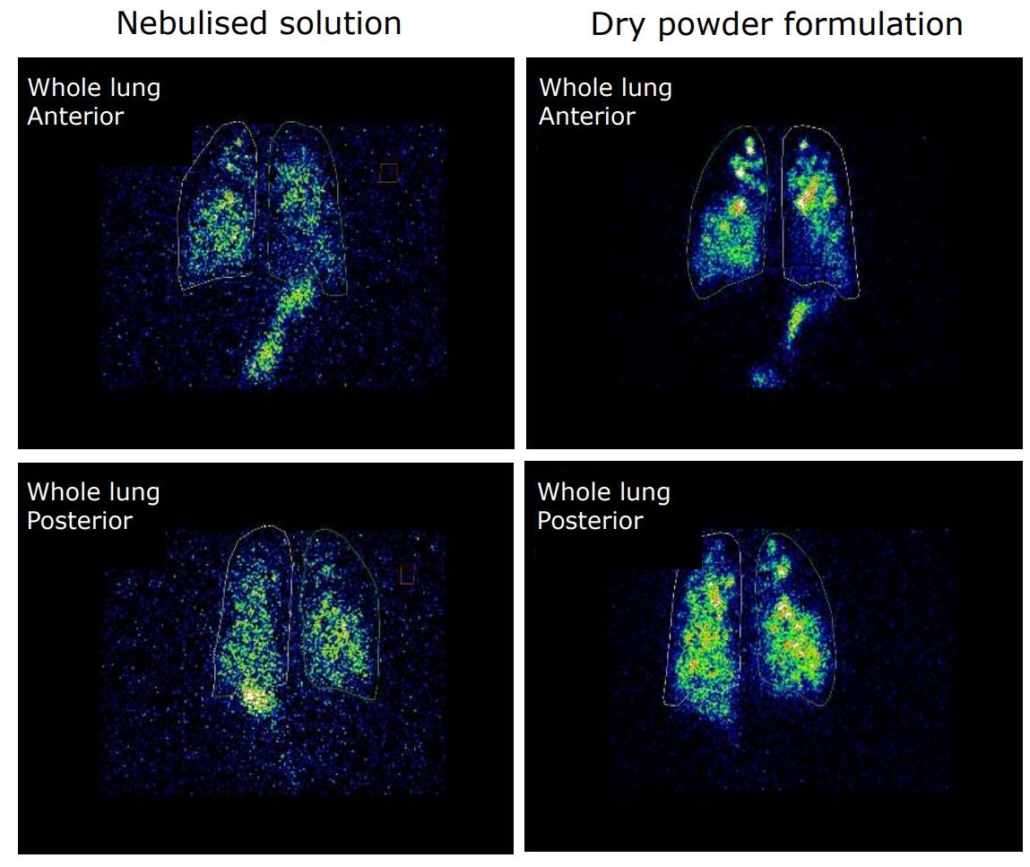
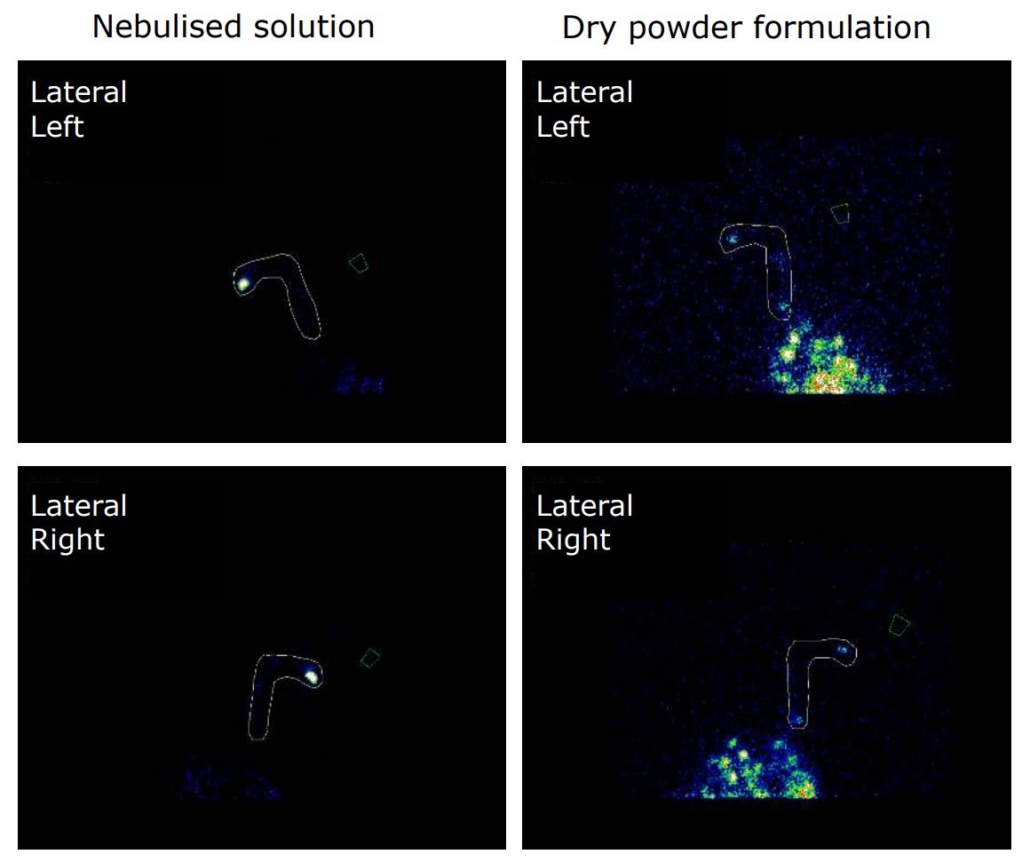
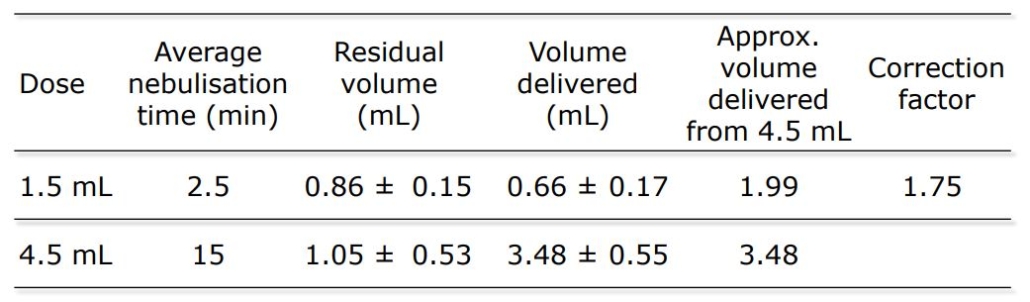
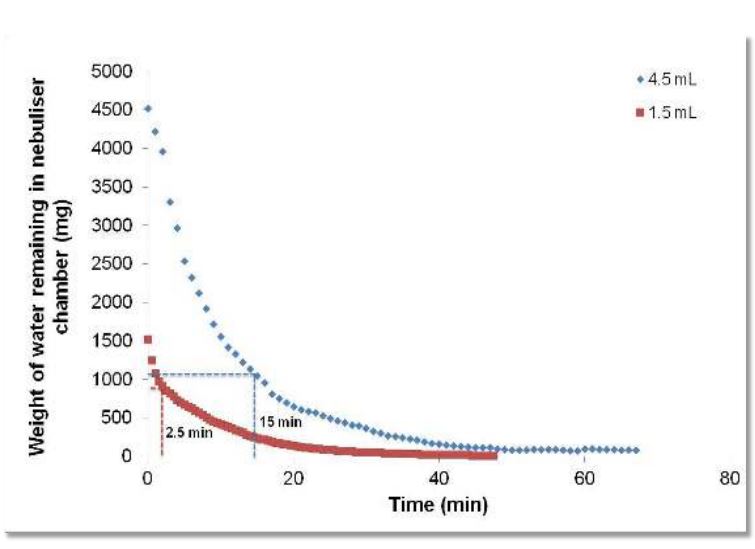
An open label, randomized two-way crossover scintigraphic study to investigate lung deposition of radiolabelled alginate oligosaccharide delivered as a dry powder and as a nebulized solution in cystic fibrosis patients
INTRODUCTION
Cystic fibrosis (CF) is a recessive genetic disease caused by mutations in CFTR leading to impaired
chloride and bicarbonate ion transport. This defect leads to accumulation of dense, intractable mucus
and impaired mucociliary clearance in the lungs, in turn causing inflammation, lung infections and tissue
obstruction.
OligoG CF-5/20 is a low molecular weight alginate oligosaccharide (Fig. 1) derived from brown algae,
comprised mainly of guluronate monomers. It has an inherent ability to bind divalent cations and has
been shown to disrupt bacterial biofilms in vitro and in animal models. OligoG can increase microbial
susceptibility towards antibiotics and antifungals in vitro [1-4]. It has also been shown to reduce mucus
viscosity in ex vivo CF sputum [5], and normalize the rheology of stagnant mucus in an ileal explant
model from CftrΔ508 mutant mice [6].
OligoG is currently in clinical development for cystic fibrosis, and has demonstrated excellent safety and
tolerability in healthy volunteers and CF patients.
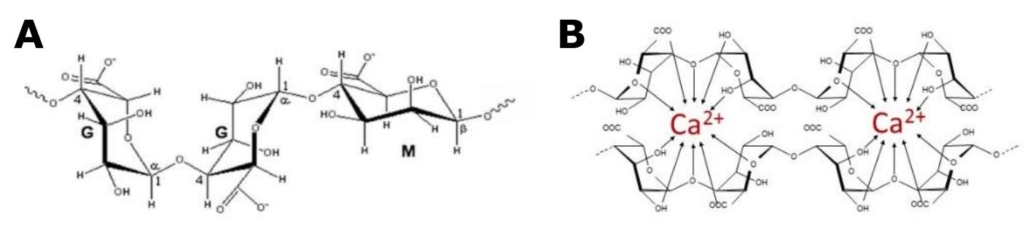
Figure 1 A: Structural composition of OligoG, showing -L-guluronate (G) and -D-manuronate
(M). At least 85% of the monomers in OligoG are G residues. B: OligoG chelating calcium